By Stephen Matthews, Ph.D.
This article is based on the dissertation of Dr. Stephen Matthews who defended his thesis at PSCOM on June 2, 2021.
TL;DR: Genetic variants can be linked to diseases through genome-wide association studies. While some variants are found in protein coding genes, many are found in non-coding regions of the genome, leaving their relevance to disease unclear. My work identified the role of a Crohn’s disease-associated SNP in a non-coding region of the genome that controlled expression of the proto-oncogene c-MYC.
Shortly after entering the atomic era and discovering DNA as the core of genetic material that passes on traits1, 1950’s films like The Fly and Them! placed irradiated and genetically mutated monsters in the film spotlight. While films like these are well-known catalysts of 20th century science fiction, science and fiction have actually grown hand-in-hand for centuries. We look back and laugh at stories where giant ants terrorize small New Mexican towns, but what made these stories impactful was their relevance to scientific understanding of the time. Today, we know that irradiation does not cause ants to grow eight feet long, but (in a less exciting reality) it can cause genetic variants that lead to disease. In fact, there are many ways genetic variants can be inherited or introduced, but not all of them result in an observable, physical change. This knowledge has real implications on human disease, and is continually expanding our understanding of genetics and molecular biology. My dissertation work at Penn State College of Medicine involved better understanding the mechanisms of how genetic variation influences normal cellular function in the intestines and its possible influence on Crohn’s disease.
First, how are genetic variations linked to disease?
Scientific understanding of genetics has long supported that genetic variation can result in changes to a person. Gregor Mendel first discovered that inheritance of some traits can be manipulated and discretely inherited. About a century later, we now know that mutations in genes like APC or BRCA2 result in high penetrance of colorectal cancer2-4 and breast cancer5-7, respectively. Importantly, while these genes are mutated in, and drive cancer progression, they also lead to mutations in other genes. Together, mutations in several genes work together to transform a normal cell to a cancerous cell. In a similar fashion, inflammatory bowel disease (IBD)* is another multifactorial disease, and results from the combination of many genetic and environmental triggers that together influence the development of IBD8. Individually these factors often contribute little to disease development, but when combined, they bring to mind the old adage “the straw that broke the camel’s back.” While the precise influence and contribution of various environmental factors is debated, there is strong evidence for genetic predisposition for IBD. Support for the role of genetics in IBD stems from several studies indicating a higher likelihood of developing IBD if first-degree relatives already have IBD9-11. That is, for many people (but not all) there is an inherited familial component to developing IBD. Identification of these individual genetic variants can be accomplished through genome-wide association studies (GWAS).
GWAS compare two populations of people, a group with the trait or disease of interest (e.g., IBD) and a group without that trait or disease (no IBD). While humans share many core traits between each other, there is still a substantial degree of genetic variation between us. Using large populations of people in GWAS can help overcome this “background” variation. Even so, the human genome is huge, and sequencing it for every single person is extensive and expensive. Often GWAS utilize marker SNPs (single nucleotide polymorphisms) to label large areas of the genome that are inherited together. That is to say, based off the A,T,C or G nucleotide identified at one spot in the genome, you can infer the status of the surrounding region. A visual representation of this can be seen in Figure 1. Here, two populations are being compared based off the presence or absence of SNP that changes a T (black) to a C (red). The more often a genetic variation is present in one population, the more likely it contributes to that trait. In Figure 1, more people in the disease group have the red SNP, therefore this SNP is associated with the disease phenotype. This does not necessarily mean the SNP you see is causative but tells you that a variant in this region may contribute to the disease of interest.

Through the use of GWAS, over 200 SNPs have been associated with IBD12, 13. While this has provided a critical insight to the disease, approximately 80% of these SNPs are not found in protein-coding sequences. The Central Dogma of biology states that DNA encodes for mRNA that makes the proteins which basically run everything in your body. However, something of which you may not be aware, is that the mRNA and proteins in your body originate from less than 2% of your genome14. The majority of your DNA does not code for proteins. Think of these non-coding sections like the font options, sizing, and spaces between words and paragraphs in a double-spaced Word document. These aspects of writing may not be directly part of the document, but are still important for your ability to read and understand the writing. In your cells, the non-coding regions of the genome influence the function of genes through recruitment of DNA binding proteins, called transcription factors. Transcription factors (TFs) are recruited to sections of DNA based off the DNA sequence, like a key fitting a lock. Much like a key, some are very specific and unique, while others are a little more promiscuous.

Once TFs bind DNA, through several steps, they either increase or decrease the expression of a nearby gene. Regions of DNA that manipulate nearby genes in this way are called “enhancers.” When enhancers are bound by their TFs, they will loop to their target genes as seen in Figure 2. These interactions bring TFs and their cofactors in close proximity to genes, facilitating easier, more rapid activation (or repression) of target genes. You can see several enhancers in Figure 2 loop to control the expression of c-MYC, along with their cofactors and TFs. In Figure 2, you will also notice four different enhancer elements (represented by purple boxes) loop with TCF/LEF, and co-factors (beta-catenin, and chromatin remodeling complexes) to increase the expression of the gene c-MYC. This interaction is complicated and contains several different co-factors, as well as inclusion of both individual enhancers spaced across this region of the genome, and a large cluster of enhancers.
Wnt-responsive enhancers and the Yochum Lab
Our lab is interested in these enhancers due to the critical nature of the signaling pathway that controls their function. The Wnt signaling pathway drives growth and proliferation of cells, which is scientifically important not only for maintaining normal intestinal function, but also for its ability to promote several diseases if inappropriately activated16. When the pathway is activated, the TCF/LEF family of TFs bound to the enhancers recruit specific co-factors and activate expression of target genes like c-MYC. The overarching hypothesis for my dissertation work was that some IBD-associated SNPs localize to enhancers controlled by Wnt signaling, and that these Wnt-responsive enhancers are deregulated in IBD to promote the disease. To address this, we looked for overlap in the location of these IBD-associated SNPs and Wnt responsive enhancers. We identified several possible targets, but I ultimately focused on the SNP rs6651252. This SNP is found in a non-coding region of the genome and is directly adjacent to the DNA sequence that TCF/LEF bind, which is important because this SNP may cause a change in how TCF/LEF bind, thereby changing the function of the enhancer (Figure 3).
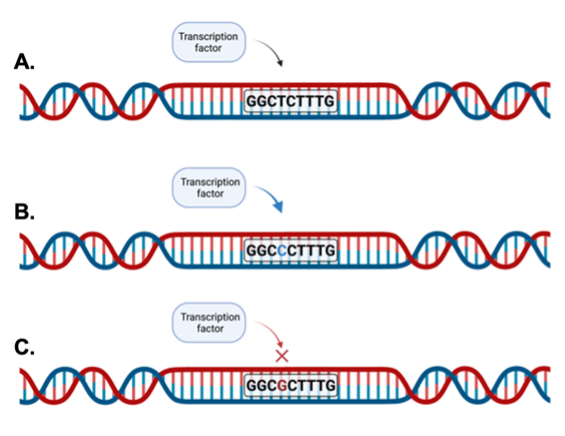
The SNP rs6651252 demarcates an enhancer that increases MYC expression
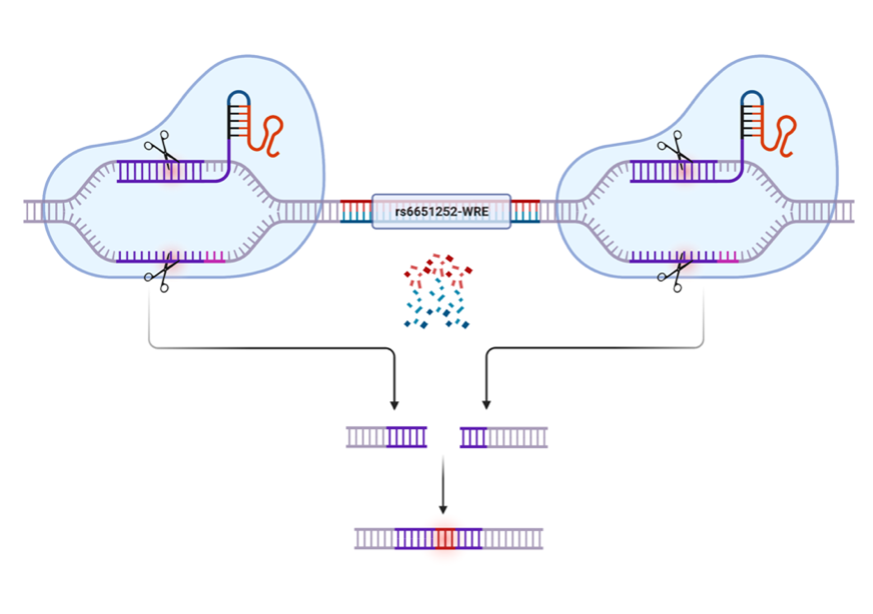
Through the work of my PhD thesis, I was able to show that the TCF/LEF family of TFs bind to the region surrounding rs6651252. Additionally, I found that the presence of the rs6651252 SNP resulted in stronger biding of the TFs. These results led me to place the sequence of DNA surrounding rs6651252 within a luciferase reporter vector, allowing me to show that the region functioned as an enhancer, increasing the amount of luciferase being produced. Luciferase is an enzyme found in the abdomen of fireflies, and can produce light. By reading the amount of light produced, I was able to determine the impact of the region I included. An enhancer elevates the amount of light emitted, while a region that does not function as an enhancer will not change the amount of luciferase being produced. Including the DNA sequence with rs6651252 in this vector increased the enhancer’s function even more, showing that the SNP increases the function of the enhancer. To determine the gene target of the enhancer, we used the gene-editing technique, CRISPR-Cas9. CRISPR uses guide RNA (gRNA) that bind a sequence of DNA and recruits Cas9, a protein that cuts DNA. As shown in Figure 4, we used this technology to cut the DNA up- and down-stream of the enhancer, resulting in its deletion. We then examined the expression of nearby potential target genes. When the enhancer was absent, the expression of c-MYC is reduced, arguing c-MYC is a target of this enhancer.
Why is this important?
c-MYC is a well-known proto-oncogene. This means the c-MYC protein is directly involved in promoting cellular growth and division, and can be deregulated to promote inappropriate, cancerous growth. While we still do not know the full downstream consequences of MYC regulation by rs6651252 or its enhancer, we have still made an important discovery in IBD research. Yet, the work continues, for there are over 200 SNPs associated with promoting IBD, and the impact of the vast majority of them still remain unclear.
So, the next time you are watching Stargate, Star Trek or a B-movie, become the life of the party by reminding everyone that irradiation and genetic variations do not cause ants to grow to the size of cars. Instead, they often result in damage that may not manifest as a disease for years or decades. Identifying these inaccuracies is only possible due to the drastic scientific advances made by scientists around the world, and these discoveries help uncover, understand, and treat diseases in an ever-improving healthcare field. The truth is more complicated than you see on the TV, but as they say in the X-files: the truth is out there!
If you are interested in reading a more in-depth and scientific explanation of my work, this is published and publicly available at: https://doi.org/10.1371/journal.pone.0212850
* This article focuses on Crohn’s disease, one of two main subtypes of IBD. If you are interested in learning more about IBD, Crohn’s disease and Ulcerative Colitis, you can do so by checking out this previous LTS article (https://lions-talk-science.org/2020/12/09/i-make-the-good-guts-go-bad-an-introduction-to-inflammatory-bowel-disease/)
All figures created in Biorender by S.M.
References:
1. Avery OT, Macleod CM, McCarty M. STUDIES ON THE CHEMICAL NATURE OF THE SUBSTANCE INDUCING TRANSFORMATION OF PNEUMOCOCCAL TYPES : INDUCTION OF TRANSFORMATION BY A DESOXYRIBONUCLEIC ACID FRACTION ISOLATED FROM PNEUMOCOCCUS TYPE III. J Exp Med. 1944;79(2):137-58. Epub 1944/02/01. doi: 10.1084/jem.79.2.137. PubMed PMID: 19871359; PMCID: PMC2135445.
2. Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet. 1992;1(4):229-33. Epub 1992/07/01. doi: 10.1093/hmg/1.4.229. PubMed PMID: 1338904.
3. Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, Koyama K, Utsunomiya J, Baba S, Hedge P. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253(5020):665-9. Epub 1991/08/09. doi: 10.1126/science.1651563. PubMed PMID: 1651563.
4. Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359(6392):235-7. Epub 1992/09/17. doi: 10.1038/359235a0. PubMed PMID: 1528264.
5. Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, Pasini B, Radice P, Manoukian S, Eccles DM, Tang N, Olah E, Anton-Culver H, Warner E, Lubinski J, Gronwald J, Gorski B, Tulinius H, Thorlacius S, Eerola H, Nevanlinna H, Syrjäkoski K, Kallioniemi OP, Thompson D, Evans C, Peto J, Lalloo F, Evans DG, Easton DF. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72(5):1117-30. Epub 2003/04/05. doi: 10.1086/375033. PubMed PMID: 12677558; PMCID: PMC1180265.
6. Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25(11):1329-33. Epub 2007/04/10. doi: 10.1200/jco.2006.09.1066. PubMed PMID: 17416853; PMCID: PMC2267287.
7. Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, Jervis S, van Leeuwen FE, Milne RL, Andrieu N, Goldgar DE, Terry MB, Rookus MA, Easton DF, Antoniou AC, McGuffog L, Evans DG, Barrowdale D, Frost D, Adlard J, Ong KR, Izatt L, Tischkowitz M, Eeles R, Davidson R, Hodgson S, Ellis S, Nogues C, Lasset C, Stoppa-Lyonnet D, Fricker JP, Faivre L, Berthet P, Hooning MJ, van der Kolk LE, Kets CM, Adank MA, John EM, Chung WK, Andrulis IL, Southey M, Daly MB, Buys SS, Osorio A, Engel C, Kast K, Schmutzler RK, Caldes T, Jakubowska A, Simard J, Friedlander ML, McLachlan SA, Machackova E, Foretova L, Tan YY, Singer CF, Olah E, Gerdes AM, Arver B, Olsson H. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. Jama. 2017;317(23):2402-16. Epub 2017/06/21. doi: 10.1001/jama.2017.7112. PubMed PMID: 28632866.
8. Connelly TM, Koltun WA. Inflammatory Bowel Disease: Pathobiology. In: Steele SR, Hull TL, Read TE, Saclarides TJ, Senagore AJ, Whitlow CB, editors. The ASCRS Textbook of Colon and Rectal Surgery. Cham: Springer International Publishing; 2016. p. 749-69.
9. Bennett RA, Rubin PH, Present DH. Frequency of inflammatory bowel disease in offspring of couples both presenting with inflammatory bowel disease. Gastroenterology. 1991;100(6):1638-43. Epub 1991/06/01. doi: 10.1016/0016-5085(91)90663-6. PubMed PMID: 2019369.
10. Ek WE, D’Amato M, Halfvarson J. The history of genetics in inflammatory bowel disease. Ann Gastroenterol. 2014;27(4):294-303. Epub 2014/10/22. PubMed PMID: 25331623; PMCID: PMC4188925.
11. Laharie D, Debeugny S, Peeters M, Van Gossum A, Gower-Rousseau C, Bélaïche J, Fiasse R, Dupas JL, Lerebours E, Piotte S, Cortot A, Vermeire S, Grandbastien B, Colombel JF. Inflammatory bowel disease in spouses and their offspring. Gastroenterology. 2001;120(4):816-9. Epub 2001/03/07. doi: 10.1053/gast.2001.22574. PubMed PMID: 11231934.
12. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Buning C, Cohain A, Cichon S, D’Amato M, De Jong D, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, Hawkey C, Hedl M, Hu X, Karlsen TH, Kupcinskas L, Kugathasan S, Latiano A, Laukens D, Lawrance IC, Lees CW, Louis E, Mahy G, Mansfield J, Morgan AR, Mowat C, Newman W, Palmieri O, Ponsioen CY, Potocnik U, Prescott NJ, Regueiro M, Rotter JI, Russell RK, Sanderson JD, Sans M, Satsangi J, Schreiber S, Simms LA, Sventoraityte J, Targan SR, Taylor KD, Tremelling M, Verspaget HW, De Vos M, Wijmenga C, Wilson DC, Winkelmann J, Xavier RJ, Zeissig S, Zhang B, Zhang CK, Zhao H, Silverberg MS, Annese V, Hakonarson H, Brant SR, Radford-Smith G, Mathew CG, Rioux JD, Schadt EE, Daly MJ, Franke A, Parkes M, Vermeire S, Barrett JC, Cho JH. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119-24. Epub 2012/11/07. doi: 10.1038/nature11582. PubMed PMID: 23128233; PMCID: PMC3491803.
13. Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, Abedian S, Cheon JH, Cho J, Dayani NE, Franke L, Fuyuno Y, Hart A, Juyal RC, Juyal G, Kim WH, Morris AP, Poustchi H, Newman WG, Midha V, Orchard TR, Vahedi H, Sood A, Sung JY, Malekzadeh R, Westra HJ, Yamazaki K, Yang SK, Barrett JC, Alizadeh BZ, Parkes M, Bk T, Daly MJ, Kubo M, Anderson CA, Weersma RK. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47(9):979-86. Epub 2015/07/21. doi: 10.1038/ng.3359. PubMed PMID: 26192919; PMCID: PMC4881818.
14. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57-74. Epub 2012/09/08. doi: 10.1038/nature11247. PubMed PMID: 22955616; PMCID: PMC3439153.
15. Fevr T, Robine S, Louvard D, Huelsken J. Wnt/beta-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol Cell Biol. 2007;27(21):7551-9. Epub 2007/09/06. doi: 10.1128/mcb.01034-07. PubMed PMID: 17785439; PMCID: PMC2169070.
16. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192-205. Epub 2012/06/12. doi: 10.1016/j.cell.2012.05.012. PubMed PMID: 22682243.
