By Laura Budurlean
All cancers exhibit some degree of genomic instability, but what if we’ve been severely underestimating how much instability there really is? Structural variants (SVs) are a hallmark of human cancer and encompass insertions, deletions, duplications, inversions, translocations, and copy number variations (Figure 1). These are all mutations that can affect large sections of the genome at once1. SVs differ from single nucleotide variations (SNVs) which only affect one, or a few base pairs of DNA. These SVs drive genomic instability and may contribute to tumorigenesis in a disproportionate way compared to SNVs which are more prevalent, but affect much smaller portions of DNA2,3. To identify SVs current technologies used in clinic involve sequencing either small targeted sections of the coding genome, called exome sequencing, or sequencing the entire human genome to more comprehensively capture SVs. Also used is karyotyping, which is a technique where entire chromosomes are examined under a microscope, or fluorescently labeled and imaged, but this provides less resolution than sequencing1,4,5.
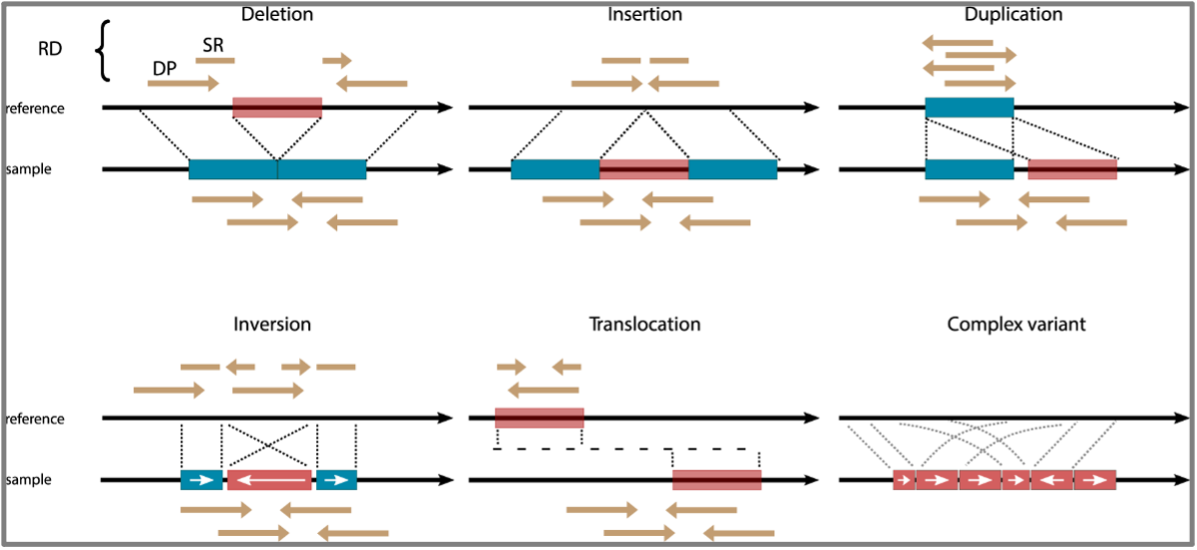
Next-generation short-read sequencing (NGS), which is a high yield sequencing method that can sequence many fragments of DNA at once, has one major disadvantage. We cannot actually see an SV; we can only use SV calling algorithms, such as LUMPY, to predict that it might be occurring1,6. Predictions of SVs are made when a patient’s genome does not align exactly to the reference genome at a particular site in the DNA. Unlike karyotyping, which focuses on very large SVs (typically over 10-20mb in length), the short reads of NGS are limited in that they cannot cover large repetitive regions or large insertions with accuracy7.
Optical genome mapping (OGM), developed by Bionano, is a technology used to detect SVs using long-reads of up to several million base-pairs to image single long pieces of DNA. Although OGM does not sequence DNA at a base pair resolution like NGS, it labels DNA with a fluorescent DNA labeling enzyme (DLE1) that creates a barcoded image of DNA which can be lined back up to a reference genome to detect discrepancies (Figure 2)8. Unlike NGS, an important distinction of Bionano is that an SV is not predicted; it is observed by direct visualization of the DNA. Because long reads can span the entirety of an SV, we can physically see when it occurs. It is similar to a geneticist examining all of a patient’s chromosomes in karyotyping, but with much better resolution because Bionano OGM can detect SVs greater than 500 bp.

Approximately two thirds of the genome is comprised of repetitive elements that can span very large regions. Sequencing over these areas is impossible with short read NGS data as there is difficulty in aligning reads correctly to the reference7,9. This is similar to proofreading a paper, but only having access to one random word at a time. Large mistakes, such as needing to delete entire sentences, would be difficult as you would never know what sentence a word belonged in. Consequently, SVs in these regions can go largely undetected. This poses a problem, as many SVs have proven to be valuable prognostic indicators in the clinic and some may be targeted by therapies that have improved patient outcome10–13.
Table 1: Genomic alterations and their clinical impact in pediatric acute lymphoblastic leukemia. Table reproduced from Kato, et al., Pediatr Int. 2018
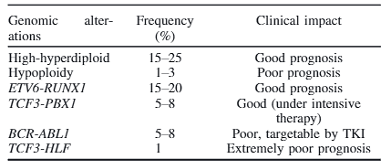
An example of this method of identification being effective is in the case of B-cell acute lymphoblastic leukemia (B-ALL), a cancer that exhibits abundant genomic structural abnormalities that can confer good or poor prognostic outcomes depending on the specific SV identified in a patient (Table 1)14. A hallmark example is the BCR-ABL1 translocation between chr9 and chr22 which forms an cancer-causing tyrosine kinase protein that is now treatable with tyrosine kinase inhibitors such as the drug imatinib15. Since many SVs are prognostic indicators (Table 1) therapy may be better personalized to the patient and comprehensive identification of SVs in the genome provides us with more opportunities to offer these targeted therapies and improve patient outcomes.
TL:DR
- Structural variations (SVs) are large, hallmark mutations in tumorigenesis.
- Many SVs remain undetectable by short read sequencing technologies.
- Bionano optical mapping uses long reads to image DNA and SVs can be physically observed.
- Detecting SVs helps develop targeted therapies and improve patient outcomes.
References
1. van Belzen IAEM, Schönhuth A, Kemmeren P, Hehir-Kwa JY. Structural variant detection in cancer genomes: computational challenges and perspectives for precision oncology. npj Precis Oncol 2021 51. 2021;5(1):1-11. doi:10.1038/s41698-021-00155-6
2. Sachidanandam R, Weissman D, Schmidt SC, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409(6822):928-933. doi:10.1038/35057149
3. Iafrate AJ, Feuk L, Rivera MN, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36(9):949-951. doi:10.1038/NG1416
4. Li Y, Roberts ND, Wala JA, et al. Patterns of somatic structural variation in human cancer genomes. Nature. 2020;578(7793):112-121. doi:10.1038/s41586-019-1913-9
5. Chen X, Bahrami A, Pappo A, et al. Recurrent Somatic Structural Variations Contribute to Tumorigenesis in Pediatric Osteosarcoma. Cell Rep. 2014;7(1):104-112. doi:10.1016/J.CELREP.2014.03.003
6. Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: A probabilistic framework for structural variant discovery. Genome Biol. 2014;15(6):6. doi:10.1186/gb-2014-15-6-r84
7. Mahmoud M, Gobet N, Cruz-Dávalos DI, Mounier N, Dessimoz C, Sedlazeck FJ. Structural variant calling: The long and the short of it. Genome Biol. 2019;20(1):1-14. doi:10.1186/S13059-019-1828-7/TABLES/2
8. Pang AWC, Lee J, Hong K, et al. Comprehensive Detection of Germline and Somatic Structural Mutation in Cancer Genomes by Bionano Genomics Optical Mapping.; 2019. Accessed October 4, 2022. https://bionanogenomics.com/wp-content/uploads/2019/09/Bionano-Genomics-Germline-Somatic-Mutations_Andy-Pang_AGBT-PH-2019.pdf
9. Tørresen OK, Star B, Mier P, et al. Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic Acids Res. 2019;47(21):10994-11006. doi:10.1093/NAR/GKZ841
10. den Boer ML, Cario G, Moorman A V., et al. Outcomes of paediatric patients with B-cell acute lymphocytic leukaemia with ABL-class fusion in the pre-tyrosine-kinase inhibitor era: a multicentre, retrospective, cohort study. Lancet Haematol. 2021;8(1):e55-e66. doi:10.1016/S2352-3026(20)30353-7
11. Rosales-Rodríguez B, Núñez-Enríquez JC, Mejía-Aranguré JM, Rosas-Vargas H. Prognostic Impact of Somatic Copy Number Alterations in Childhood B-Lineage Acute Lymphoblastic Leukemia. Curr Oncol Rep. 2021;23(1):1-11. doi:10.1007/s11912-020-00998-5
12. Stanulla M, Cavé H, Moorman A V. IKZF1 deletions in pediatric acute lymphoblastic leukemia: Still a poor prognostic marker? Blood. 2020;135(4):252-260. doi:10.1182/blood.2019000813
13. Bernt KM, Hunger SP. Current concepts in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia. Front Oncol. 2014;4 MAR(March):1-21. doi:10.3389/fonc.2014.00054
14. Kato M, Manabe A. Treatment and biology of pediatric acute lymphoblastic leukemia. Pediatr Int. 2018;60(1):4-12. doi:10.1111/ped.13457
15. Igwe IJ, Yang D, Merchant A, et al. The presence of Philadelphia chromosome does not confer poor prognosis in adult pre-B acute lymphoblastic leukaemia in the tyrosine kinase inhibitor era – a surveillance, epidemiology, and end results database analysis. Br J Haematol. 2017;179(4):618-626. doi:10.1111/bjh.14953
